

三菱ケミカル、慶大および日本IBMは、光機能性物質のエネルギーを高い精度で求めるために、[A]スピン保存量子回路の設計指針と、[B]励起状態の新しい計算方法「制約条件自動調整変分量子固有値法(VQE/AC法)」を開発しました。これら[A][B]を組み合わせ、日本初のゲート型商用量子コンピューター「IBM Quantum System One」※4上で、フェノールブルー色素の光吸収と熱失活に関わる構造でのエネルギー計算を行った結果、基底状態・励起状態のエネルギーをわずか2 kcal/molの誤差範囲で求めることに成功しました。
この研究成果は、光機能性物質の理解と設計に不可欠な基底状態・励起状態上での構造最適化への道を開くことが期待されます。
三菱ケミカル、慶大および日本IBMは、今後も、幅広い材料開発に用いるための量子コンピューターの技術確立を進めていきます。
※1:三菱ケミカルグループは、三菱ケミカルグループ株式会社とそのグループ会社の総称です。
※2:慶大と日本IBMが2018年5月に慶大理工学部に開設した最先端の量子コンピューター研究拠点です。IBM が開発した最先端の量子コンピューターのクラウド利用を可能とするアジア初の IBM Quantum Hubであり、産学共同の研究拠点として三菱ケミカルは発足メンバーとして参画しています。
※3:掲載論文のURL=https://doi.org/10.1038/s41524-023-00965-1
※4:国立大学法人東京大学とIBMで締結された「Japan–IBM Quantum Partnership」に基づき、2021年7月から「新川崎・創造のもり かわさき新産業創造センター(KBIC)」にて稼働開始した設備です。
以上
【本研究のポイント】
・量子コンピューターによる光機能性物質の高精度な計算のために、スピン保存量子回路とVQE/AC法※5を開発
・スピン保存量子回路を用いることで、エネルギーの計算誤差を最小化
・VQE/AC法を用いることで、従来法で必要だった事前パラメタ調整を不要とした
・光機能性物質の理解と設計に不可欠な構造最適化計算への応用が期待できる
 図1. フェノールブルー色素のFC構造(H,C,N,O原子を白,灰,青,赤色で記載)とCI構造(緑色で記載)
図1. フェノールブルー色素のFC構造(H,C,N,O原子を白,灰,青,赤色で記載)とCI構造(緑色で記載)
【背景】分子の光機能の理解の深化や合理的設計には、光吸収・発光の波長や強度、発光せずに熱失活してしまう確率などを高精度に求めることが必要です。これらの量を求めるには、各現象が最も起こりやすくなる分子構造(FC構造※6やCI構造※7)における基底状態※8と励起状態※9のエネルギーの定量的な計算が不可欠です。現在、励起状態の計算にはTDDFT法※10が最も広く用いられていますが、CI構造のように基底状態と励起状態のエネルギー差がない、または小さい場合、TDDFT法ではエネルギーを正しく求めることができず、代わりに多参照の計算方法※11を用いる必要があることが知られています。しかし、多参照の計算を複雑な分子に適用すると、膨大な計算コストがかかるという問題がありました。
この問題の解決の切り札として注目を集めているのが量子コンピューターです。量子コンピューター上で励起状態を計算するためには、基底状態と励起状態を表現できる量子回路※12と励起状態計算を行うためのコスト関数を適切に設計しなければなりません。量子コンピューターを用いた計算には、必ず誤差※13が含まれるため、誤差を最小におさえる量子回路の設計が重要課題でした。また、励起状態計算に適切なコスト関数が、分子構造によって異なるため、すべての分子構造に共通して適用可能な計算方法が求められていました。
【今回の成果】
本研究では、[A]スピン多重度を保存する量子回路(スピン保存量子回路)の設計指針と[B]コスト関数を用いない励起状態計算法(VQE/AC法)を開発することで、従来よりも小さな計算誤差での励起状態計算に成功しました。
[A] スピン多重度を保存する量子回路の設計
量子回路の設計において、基底状態と励起状態のスピン多重度を保存するという条件を課しました。例えば、2つの分子軌道(HOMOとLUMO※14)に2つの電子を含む一重項状態は、図2aに示す3つの電子配置の重ね合わせで表されます。この3つの電子配置を過不足なく表現する回路(図2b)を設計することで、誤差※13の原因の一つである電子数やスピン多重度の異なる成分の混入を防いでいます。実際に、スピン保存量子回路は、他の設計指針に基づいた回路よりも小さい計算誤差でエネルギーを計算できることを明らかにしました。
 図2. スピン保存量子回路の設計
図2. スピン保存量子回路の設計
(a) 2つの軌道(HOMO、LUMO)に2電子を含む一重項の電子配置。(b) 3種の一重項電子配置の重ね合わせを表現する量子回路。2本の線q0とq1は、2つの量子ビットを表す。四角で表した量子ビット演算に含まれるパラメタq0とq1を古典コンピューターで最適化することで、エネルギーを求めます。
[B] コスト関数を用いない励起状態計算法(VQE/AC法)の開発
従来の励起状態計算(VQD法※15)では、図3のコスト関数に含まれるパラメタbとg を事前に調整した上で、コスト関数からエネルギーを求めていました。事前調整が必要なパラメタbとg の適切な値は、分子構造に依存するため、VQD法を用いてポテンシャルエネルギー局面※16を記述するのは困難でした。しかしながら、スピン保存量子回路を用いることにより、g をゼロに固定することが可能になります。さらに、 をゼロに近い値にするという制約条件を課した最適化手法を用いることで、b の事前調整も回避することができました。このように、励起状態が満たすべき条件を制約とする最適化計算によって励起状態を計算する方法を、「制約条件自動調整変分量子固有値法(VQE/AC法)」と名付けました。
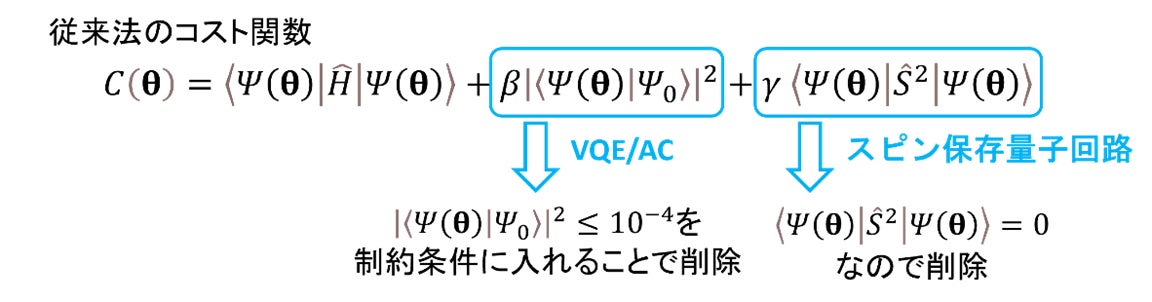
図3. 従来法におけるコスト関数 とVQEAC法の概念
実際にスピン保存量子回路とVQE/AC法を組み合わせた多参照計算の一種であるCASSCF法※17を用いて、フェノールブルー色素の基底状態・励起状態を、新川崎・創造のもり かわさき新産業創造センターに設置されたゲート型量子コンピューターIBM Quantum System One上で計算したところ、FC構造、CI構造いずれの場合も、計算誤差をわずか2 kcal/molに抑えてエネルギーを求めることに成功しました。
【今後の展望】
VQE/AC法は励起状態計算のために開発した方法ですが、制約条件を変えることで、励起状態に限らず、様々な状態の計算に応用することができます。また、VQE/AC法を用いることで、任意の分子構造の基底状態・励起状態のエネルギーを同一の計算条件で求められるようになったため、将来的には安定構造や状態間の交差点の構造最適化への応用が期待できます。
【用語解説】
※5:VQE/AC法
本研究で開発したVariational quantum eigensolver under automatically-adjusted constraintsの略。
※6:FC構造
フランク=コンドン(Frank-Condon)構造の略。基底状態において最も安定な構造。光吸収が最も起こりやすい構造でもある。
※7:CI構造
円錐交差(Conical intersection)構造の略。基底状態と励起状態のエネルギーが一致する構造。励起状態からの失活が最も起こりやすい構造である。
※8:基底状態
最もエネルギーの低い電子状態。
※9:励起状態
基底状態よりもエネルギーが高い電子状態。基底状態と励起状態のエネルギー差に等しいエネルギーを持つ光を吸収することで、到達することができる。
※10:TDDFT法
時間依存密度汎関数(Time dependent density functional theory)法の略。
※11:多参照の計算方法
基底状態や励起状態を複数の電子配置で記述する方法。多参照配置間相互作用法(MRCI)、多参照摂動論(MRPT)、多参照自己無撞着場法(MRSCF)など様々なバリエーションがある。
※12:量子回路
量子ビットの量子状態を制御する量子ゲートや測定を組み合わせたもの。
※13:誤差
統計誤差と量子デバイスのノイズに由来する誤差をまとめて誤差と表現している。
※14:HOMOとLUMO
電子占有軌道のうち、最も不安定な分子軌道をHOMO、空軌道のうち、最も安定な軌道をLUMOという。
※15:VQD法
Variational quantum deflationの略。
※16:ポテンシャルエネルギー局面
構造変化に対するエネルギー変化をプロットした図。
※17:CASSCF法
Complete active space self-consistent field法の略。電子配置の重みと、電子配置を構成する分子軌道を同時に最適化する方法。MRSCF法の一種。
<共著者リスト>
・慶應義塾大学 :後町 慈生、畑中 美穂、稲垣 泰一
・三菱ケミカルグループ :高 玘、小林 高雄、菅野 志優
・IBM Research-Tokyo :中村 肇
